 Facebook
Facebook Google
Google GitHub
GitHub Linkedin
LinkedinAI Creates Better-Performing Batteries at the Atomic Scale
Argonne National Laboratory and the University of Michigan have collaborated to use AI, machine learning, and supercomputers to evaluate potential battery materials.
Pursuing next-generation battery technologies has reached the point where artificial intelligence meets atomic-scale materials science. Two leading research institutions, Argonne National Laboratory (ANL) and the University of Michigan (UM), have emerged as pioneers in developing AI-powered approaches to design and evaluate battery materials molecule by molecule and atom by atom.

Argonne National Laboratory’s Aurora system provides advanced AI, simulation, and data analysis in the search for battery materials. Image used courtesy of Argonne National Laboratory
Finding Battery Materials, Step by Step
Decades of incremental progress, mostly through serendipitous breakthroughs, have characterized the traditional approach to discovering battery materials. Most battery materials currently used were found between 1975 and 1985. These same materials are relied on with small, incremental changes to improve battery performance.
A battery’s performance depends on a range of complex interactions across multiple dimensional scales, from atomic arrangements of atoms to the macroscopic behavior of the device.
When AI and Atomic-Scale Materials Science Converge
Pharmacologists estimate there could be as many as 1060 possible molecular compounds. To investigate this astronomical number, scientists have moved to AI modeling to predict properties of candidate molecules to create new drugs. Now, battery researchers are using the same types of tools to develop battery material candidates.
Argonne Lab and the University of Michigan have recognized that artificial intelligence, combined with high-performance computing and advanced characterization techniques, can bridge the enormous gap between theoretical possibilities and experimental realities. Their approaches leverage complementary methods: foundation models trained on molecular data, autonomous discovery platforms, and quantum mechanical simulations that operate at the atomic scale.
This represents a paradigm shift from traditional trial-and-error methods to precision-guided materials discovery, with the goal of developing safer, more efficient, and sustainable energy storage solutions.
University of Michigan's Foundation Model Revolution
The University of Michigan has pioneered chemical foundation models—massive AI systems specifically designed for materials discovery. Their work, supported by the Department of Energy's INCITE program, has produced some of the most sophisticated molecular foundation models, trained on datasets containing billions of molecules.
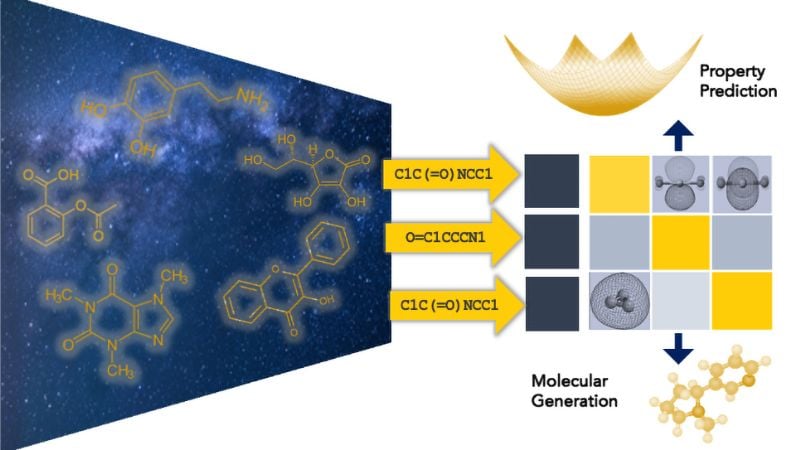
The foundational model maps the molecular structures to predict their properties and design molecules with desired properties. Image used courtesy of the University of Michigan/Anoushka Bhutani
The team's approach draws inspiration from the success of large language models like ChatGPT but adapts these concepts for chemistry. Using 200,000 node hours on Argonne's Polaris supercomputer, the Michigan team has built what they describe as the largest molecular foundation model to date, pre-trained on 2 billion molecules. This model focuses on small organic molecules composed primarily of carbon, hydrogen, oxygen, and nitrogen—the building blocks of battery electrolytes. The foundation model's power lies in its ability to predict multiple properties simultaneously, from conductivity and melting point to flammability and electrochemical stability.
Molecular Representation and Chemical Understanding
A crucial innovation in the UM work involves the development of sophisticated molecular representation systems. The team employs SMILES (Simplified Molecular Input Line Entry System), a widely used method for representing molecular structures as text strings, combined with their novel SMIRK tool that improves how the model processes these structures. This approach allows AI to learn from billions of molecules with extremely high precision and consistency.
Recent advances in molecular representation have further enhanced these capabilities. Researchers have developed hybrid methods like SMI + AIS (SMILES + Atom-In-SMILES), incorporating local chemical environment information into individual models. These enhanced representations significantly improve molecular design tasks, with 7% better binding affinity and 6% increased synthesizability compared to standard approaches.
Expanding to Solid-State Materials
Building on their success with molecular systems, the Michigan team is now leveraging Argonne’s new Aurora exascale system to develop a second foundation model focused on molecular crystals—the building blocks of battery electrodes. This expansion represents a significant leap in scope, moving from liquid electrolyte components to solid-state materials that form batteries’ active components.
The crystal-focused foundation model addresses one of the most challenging aspects of battery materials design: the complex relationship between atomic structure and macroscopic properties in solid materials. Unlike molecules, crystals exhibit long-range order and collective behaviors that emerge from atomic-scale interactions. The ability to predict these properties accurately using AI could transform electrode materials.
Argonne's Autonomous Discovery Ecosystem
While the University of Michigan focuses on developing foundational AI models, Argonne has created a comprehensive ecosystem for autonomous materials discovery. At the heart is Polybot, an AI-driven robotic laboratory housed at the Center for Nanoscale Materials. Polybot represents the convergence of artificial intelligence, robotics, and materials science in a fully autonomous experimental platform that runs experiments based on AI-driven decisions.

The Polybot in action. Image used courtesy of Argonne National Laboratory via University of Chicago
Polybot's capabilities extend far beyond simple automation. The system employs importance-guided Bayesian optimization to navigate complex, high-dimensional parameters. In recent demonstrations, Polybot successfully explored a 7-dimensional processing space for electronic polymer thin films, achieving remarkable results, including transparent conductive films with conductivity exceeding 4,500 S/cm.
Polybot’s autonomous nature addresses a fundamental limitation of traditional materials research: the sheer number of possible experimental conditions. Polybot's AI-guided approach enables systematic exploration of millions of parameter spaces with unprecedented efficiency.
The system's sophistication is evident in its experimental methodology. Polybot employs statistical methods to ensure data repeatability, implements multi-objective optimization algorithms, and can concurrently optimize multiple material properties. This comprehensive approach has enabled the platform to screen 90,000 material combinations in weeks—a task that would typically require months using manual methods.
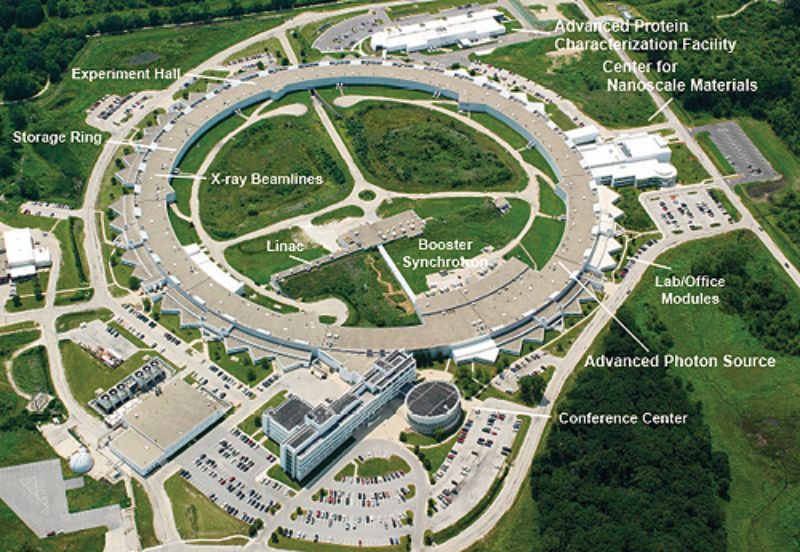
The Advanced Photon Source. Image used courtesy of ANL
The platform's connection to Argonne's Advanced Photon Source (APS) provides particularly valuable capabilities. The APS generates powerful X-ray beams that can probe materials at the atomic scale, enabling unprecedented insight into structure-property relationships. Recent work has used these capabilities to study complex interactions inside batteries during operation, revealing atomic-scale mechanisms that govern performance and degradation.
Supercomputing at Exascale and Beyond Lithium-Ion Technologies
The success of these AI-driven approaches stems partly from the synergistic collaboration between institutions and the sharing of computational resources. The University of Michigan's work extensively utilizes supercomputing resources at Argonne's Leadership Computing Facility, while Argonne researchers benefit from theoretical frameworks developed at Michigan. The Aurora exascale system at Argonne represents a quantum leap in computational capability, using the Polaris supercomputing facility that can perform a billion-billion calculations per second. This unprecedented computing power enables training ever-larger foundation models and exploring previously intractable chemical spaces.
The foundations being established extend far beyond current lithium-ion technologies. Recent work by Microsoft and Pacific Northwest National Laboratory, leveraging similar AI approaches, successfully identified a new material that could reduce lithium use in batteries by up to 70%. The AI-driven process narrowed down 32 million potential materials to 18 promising candidates in less than a week—a screening process that could have taken more than two decades using traditional methods.
Challenges and Future Directions
Despite remarkable progress, significant challenges remain in AI-driven materials discovery. Battery systems’ complexity, with their multiple interacting components and multi-scale phenomena, continues to pose obstacles.
One persistent challenge involves ensuring the quality and reliability of training data for AI models. This data must be obtained not only from successful trials but also from unsuccessful ones. This emphasis on comprehensive data collection, including negative results, is crucial for developing robust AI models. The gap between computational predictions and experimental validation also remains significant.
The convergence of AI and atomic-scale materials science at Argonne and Michigan represents more than incremental progress—it signifies a fundamental transformation in how materials are discovered, designed, and optimized. The ability to systematically explore chemical interactions, predict properties from atomic structure, and autonomously optimize processing conditions opens unprecedented opportunities for energy storage advancement.











