 Facebook
Facebook Google
Google GitHub
GitHub Linkedin
LinkedinAI Allows Examination of More Than 31 Million New Materials
A new database contains millions of never-before-existing materials that could provide breakthroughs for lithium-ion batteries.
The ability to use artificial intelligence (AI) to computationally predict the structure and performance of new combinations of atoms into previously unknown compounds has become a major part of materials engineering.
Combinations of atoms. Image used courtesy of Matterverse.ai
For example, in 2021, AlphaFold, an open-access, searchable database with more than 200 million protein structure predictions, was made available to the global scientific community allowing a protein’s 3D structure to be predicted from its amino acid sequence. AlphaFold was quickly found to be useful in areas including drug discovery, vaccine development, food security, bioinformatics, and ecology.
Now, a similar type of AI-based computational tool to calculate the structure and dynamic properties of any material has been made available by a nanoengineering team at the University of California-San Diego. The team, spearheaded by professor Shyue Ping Ong, has created an algorithm that has been used to develop a database of 31 million yet-to-be-synthesized materials along with their properties as predicted by machine learning algorithms.
Known as M3GNet, the algorithm determines the properties of a material based on the arrangement of its atoms. The database called matterverse.ai was released in February 2022 and has been used by academic and industry researchers to help find new materials in various applications.
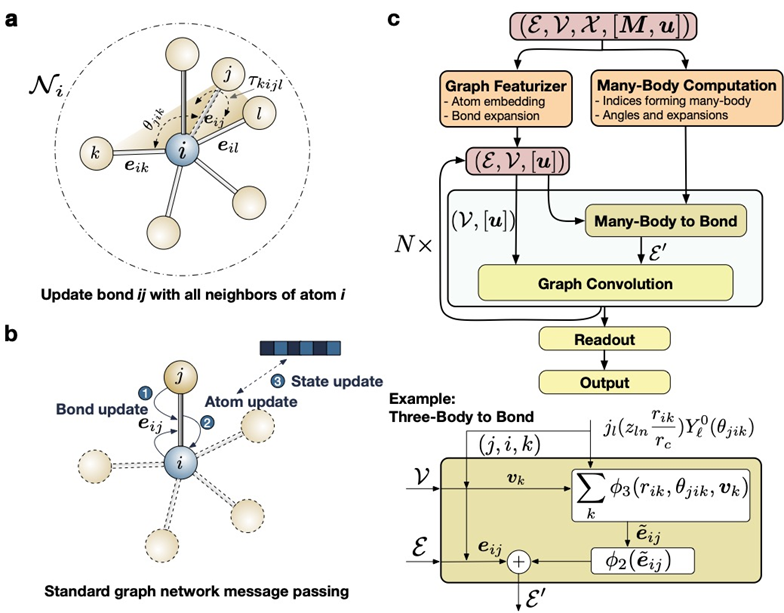
Illustration of the M3GNet algorithm. Image used courtesy of UC San Diego, Jacobs School of Engineering
Predicting the Properties of Materials for Better Batteries
To predict the properties of materials from combinations of atoms, the team used the mathematical models embodied in graph neural networks and many-body interactions to allow deep learning of architecture. A high degree of accuracy across all of the elements of the periodic table was observed from the modeling.
A huge database of existing materials and their energies, forces, and stresses were used to train the AI portion of the modeling. As a result, the M3GNet interatomic potential (IAP), can predict the forces and energies in any collection of atoms. More than 31 million materials have been virtually created, and of those, more than a million have been predicted to be stable.
Professor Ong and his team are using M3GNet to research new and previously unknown materials that can be used for higher energy-density electrodes and more effective electrolytes for lithium-ion batteries.
Battery Systems for Electric Vehicles
As electric vehicles (EVs) become more mainstream, there is increasing urgency to develop battery systems that can charge more quickly. While present-day DC fast chargers can replenish the charge in an EV battery in as little as 30-40 minutes, there is an interest in reducing that time to the 5 minutes it takes to fuel a traditional gasoline-powered car at a gas pump.
In an EV with lithium-ion batteries, one of the limiting factors in how quickly the battery can be charged is how fast lithium ions can diffuse through the electrodes and electrolyte. The UC team has found that M3GNet can be used to predict the diffusion of lithium through battery materials with a high degree of accuracy.
The AI-based prediction system can be used to evaluate large numbers of never-before-tried arrangements of atoms with the hope of finding a combination that will allow faster, safer charging.
The team has released the framework of the algorithm as an open-source Python code on Github to promote the use of M3GNet, and plans to integrate the model as a tool in commercial materials simulation packages.











